Marfan症候群(マルファン症候群)
- 結合織の異常に関連する遺伝性疾患であり、主に心血管系、眼、骨格に多様な症状を示す。
- 1896年にMarfanによって初めて報告された。
- 発生率は約5千人から1万人に1人とされており、患者の約20〜30%では新規の突然変異が原因で発症する。
- 心血管系では、大動脈弁輪拡張症や上行大動脈解離が主要な合併症であり、これが死因となることが多い。大動脈基部の拡張は進行性であり、大動脈径が50mmを超えると破裂のリスクが高まるため、外科手術が推奨されるクラスⅠに該当する。また、妊娠を希望する女性では40mm以上での治療が推奨されるクラスⅡaに分類される。
- 眼に関しては、水晶体の脱臼や扁平角膜、眼軸の異常延長などが特徴的である。
- 骨格系では、長身や痩身、長い手足、くも状指、胸郭変形、脊柱側弯などの特徴が見られる。これらの徴候は診断の重要な基準となる。
- 特に腰仙部硬膜嚢拡張症は重要な所見で、腰仙椎レベルで硬膜嚢の拡大や椎体後縁の陥凹が確認される。腰痛、神経症状、そして髄液漏れを引き起こすことがある。
- 他の類似する結合織病との鑑別が必要である。これにはロイス・ディーツ症候群、Shprintzen-Goldberg症候群、血管型エーラス・ダンロス症候群などが含まれる。これらの疾患も臨床像が似ているため、詳細な臨床所見の検討および遺伝子診断が不可欠である。
症例 50歳代男性 中等度の労作時に息切れ(Marfan症候群と診断されている)
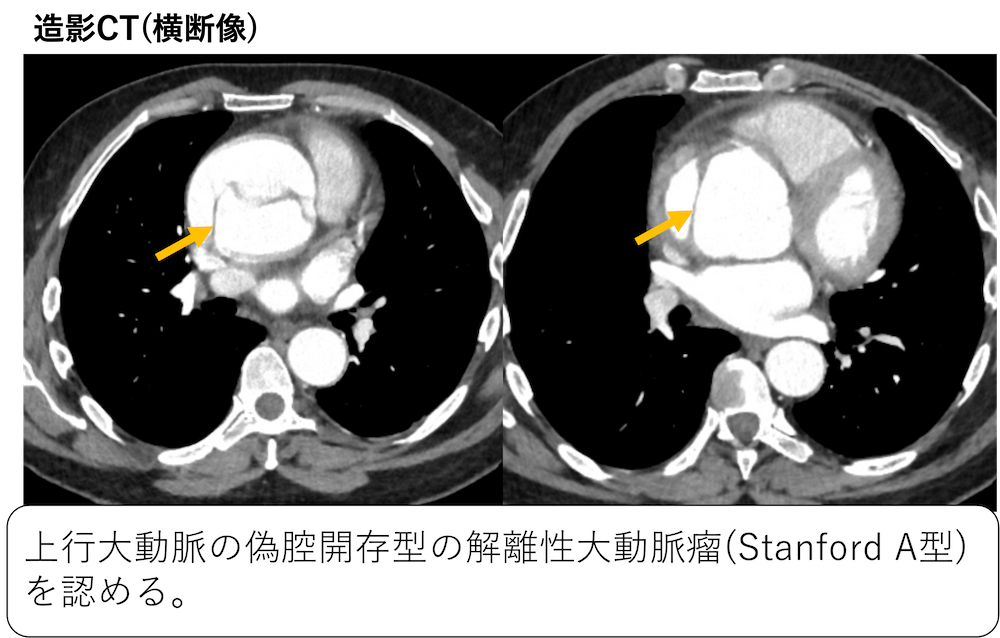
引用:radiopedia
上行大動脈の偽腔開存型の解離性大動脈瘤(Stanford A型)、大動脈弁輪拡張症を認めています。
腰仙部硬膜嚢拡張症の画像所見
- 腰仙部硬膜嚢拡張症は腰仙椎レベルで硬膜嚢の拡大や椎体後縁の陥凹、脊柱管の拡張、仙骨の菲薄化、仙骨孔の拡張を認める。CTやMRI(特にT2WI)で確認できる。
症例 10歳代女性 Marfan症候群と診断されている。
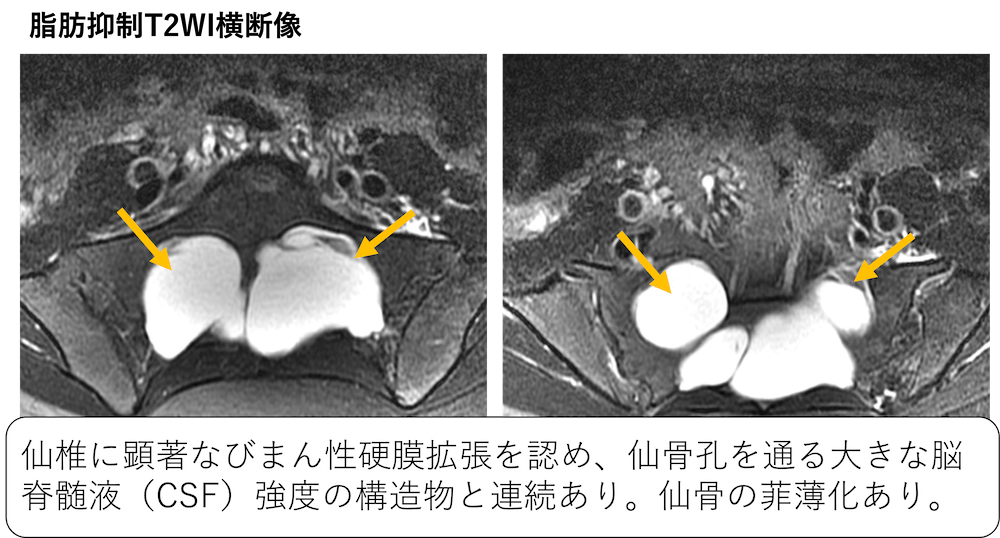
引用:radiopedia
仙椎に顕著なびまん性硬膜拡張を認め、仙骨孔を通る大きな脳脊髄液(CSF)強度の構造物と連続あり。仙骨の菲薄化あり。腰仙部硬膜嚢拡張症の所見です。
参考文献:
- 臨床放射線, Vol.67 No.11 2022, P1310
- 画像診断別冊KEY BOOKシリーズ, 心臓・血管疾患の画像診断, P270~73
- Lerner, T. J., Korf, B. R., & Dietz, H. C. (2007). Marfan syndrome. New England Journal of Medicine, 347(19), 1568-1574.
- Habermann, T. M., & Gertz, M. A. (2001). Marfan’s syndrome: The expanding phenotype. Blood, 98(4), 987-994.
- Ahn, N. U., Sponseller, P. D., Ahn, U. M., Nallamshetty, L., & Rose, P. S. (2000). Dural ectasia in the Marfan syndrome: MR and CT findings and criteria. Genetics in Medicine, 2(3), 173-179.
ご案内
腹部画像診断を学べる無料コンテンツ
4日に1日朝6時に症例が配信され、画像を実際にスクロールして読影していただく講座です。現状無料公開しています。90症例以上あり、無料なのに1年以上続く講座です。10,000名以上の医師、医学生、放射線技師、看護師などが参加中。
胸部レントゲンの正常解剖を学べる無料コンテンツ
1日3分全31日でこそっと胸部レントゲンの正常解剖の基礎を学んでいただく参加型無料講座です。全日程で簡単な動画解説付きです。
画像診断LINE公式アカウント
画像診断cafeのLINE公式アカウントで新しい企画やモニター募集などの告知を行っています。 登録していただくと特典として、脳の血管支配域のミニ講座の無料でご参加いただけます。