Huntington病(Huntington’s disease:HD/ハンチントン病)とは?
- 舞踏運動を主体とする不随意運動と認知症や性格変化などの精神症状を主症状とする神経変性疾患。
- 第4染色体短腕4p16.3上のIT15領域におけるCAGリピートの異常伸長を原因とするポリグルタミン病であり、常染色体優性遺伝する。
- わが国での有病率は10万人当たり約0.5人。
- 成人期、特に30~40歳台に発症することが多いが、世代が経過するごとにCAGリピートが蓄積する表現促進現象があり、発症時期が若年化する。
- 古典的三徴は舞踏病アテトーゼ、認知機能障害、精神症状。
- 約10%の症例は20歳未満に発症する若年型HD(Westphal variant)で、パーキンソニズムを呈する急速進行型であり、舞踏運動はまれである。
- 病理では、皮質-線条体-視聴回路に神経細胞脱落がみられる。
Huntington病(Huntington’s disease:HD)の画像所見
- 基本的な画像所見は尾状核、被殻の萎縮。T2WIで低or高信号を認めることがある。病理での神経細胞の脱落と、グリオーシスや鉄沈着を反映したもの。
- これらの萎縮は症状の発現に数年先行して出現し、尾側から頭側、背側から腹側へと進行する。
- 尾状核頭部の萎縮による側脳室前角の拡大は成人、小児症例いずれも認められる最も特徴的な所見であるが、早期では目立たないことがある。尾状核体部の萎縮は、冠状断像での、尾状核の外側への膨隆の消失と直線化してとらえられる。
- 一方、MRIやSPECTにおける被殻の萎縮や血流の低下は早期から認められる所見で、発症前の診断に有用と報告されている。
症例 40歳代男性 上肢舞踏病およびジストニア運動
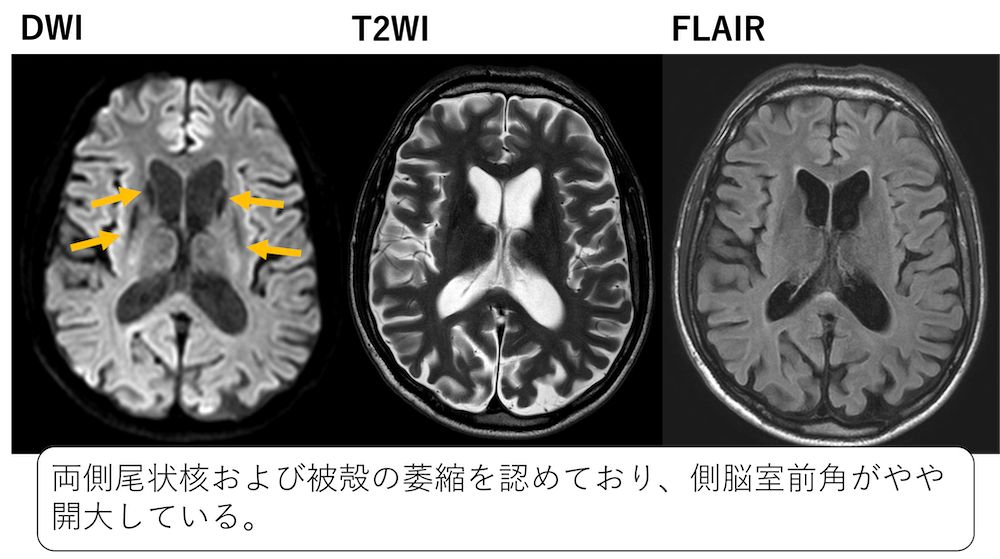
引用:radiopedia
両側尾状核および被殻の萎縮を認めており、側脳室前角がやや開大しています。
Huntington病と診断されました。
Huntington病の鑑別診断
両側基底核の萎縮やMRI異常信号を呈する疾患が鑑別となり
- 神経有棘赤血球症(neuro-acanthocytosis:nac)
- 脊髄小脳変性症17(spinocerebellar ataxia 17:SCA17)、
- GM1 gangliosidosis type 3.Wilson病(Wilson’s disease:WD)、
- Leigh脳症
- Wilson病など
特に神経有棘赤血球症は画像所見のみならず、舞踏様運動、認知機能障害などの症状があり、類似した臨床経過を呈しうる。そのため有棘赤血球や筋萎縮の有無など理学所見を含めた総合的な評価が必要。
関連記事:大脳基底核とは?機能と障害されたときに起こることまとめ
参考文献:
- 臨床画像 Vol.35 No.2 2019 P248
- 新 頭部画像診断の勘ドコロ P254
ご案内
腹部画像診断を学べる無料コンテンツ
4日に1日朝6時に症例が配信され、画像を実際にスクロールして読影していただく講座です。現状無料公開しています。90症例以上あり、無料なのに1年以上続く講座です。10,000名以上の医師、医学生、放射線技師、看護師などが参加中。
胸部レントゲンの正常解剖を学べる無料コンテンツ
1日3分全31日でこそっと胸部レントゲンの正常解剖の基礎を学んでいただく参加型無料講座です。全日程で簡単な動画解説付きです。
画像診断LINE公式アカウント
画像診断cafeのLINE公式アカウントで新しい企画やモニター募集などの告知を行っています。 登録していただくと特典として、脳の血管支配域のミニ講座の無料でご参加いただけます。